
March 2021: MaizeGDB qTeller now has data for version 5 of B73 and the NAM Founders! Select either "B73v5 [NAM Consortium]" or "NAM Founders [NAM Consortium]" under the drop-down menus above.
October 2019: MaizeGDB qTeller has a new look! Now we can compare expression and abundance under the menu 'Compare RNA & Protein'.
Questions or suggestions? Please let us know on the Contact page.
 Welcome to an updated version of qteller! Compare RNA & Protein
Welcome to an updated version of qteller! Compare RNA & Protein
Welcome to qTeller for  !
!
qTeller is a comparative RNA-seq expression platform to compare expression across multiple data sources in a user-provided gene list or genomic interval, or to visually compare expression between two genes. qTeller has been used extensively in maize and other research.
qTeller has four tools:
Genes in an Interval
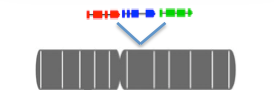
Select a chromosomal interval and expression data sources to compare the expression of all the genes in your interval.
Genes by Name

Similar to Genes in an Interval except you provide a list of genes as input instead of a chromosomal interval.
Visualize expression
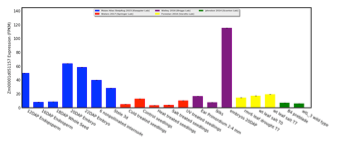
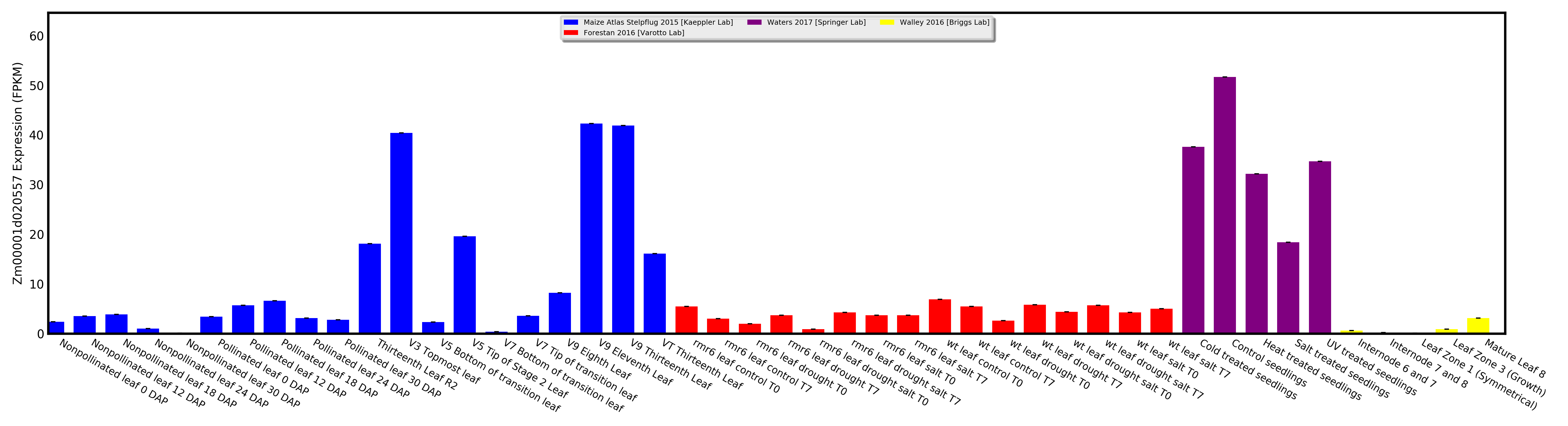
Visualize expression for one gene as a bar chart, or visually compare expression between two genes as a scatterplot.
Compare RNA & Protein
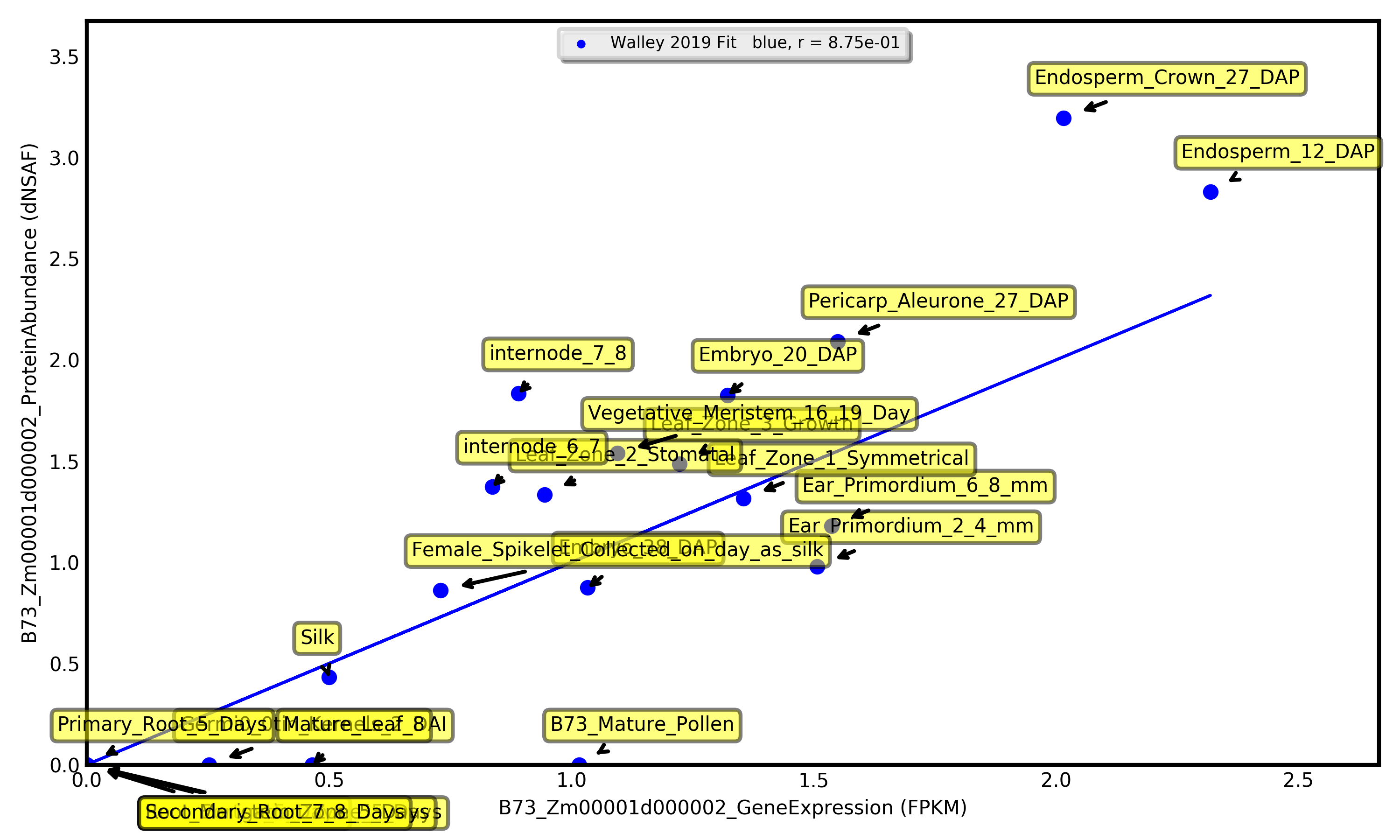
Visualize expression for one gene as a bar chart, or visually compare expression between two genes as a scatterplot.
Getting started
Getting started
1. Select your tool of interest at the top navigation bar (Genes in an interval, Genes by name, Visualize expression, or Compare RNA & Protein), then select your genome of interest from the tool dropdown menu.
2. Then follow the directions on the tool page!
Frequently Asked Questions (FAQ):
What mapping pipeline does MaizeGDB qTeller use?
For high-throughput mapping of hundreds of SRA runs, we have deviated from the Classic qTeller pipeline by using the STAR mapping algorithm instead of GSNAP. However, we use Cufflinks to count FPKM, just as Classic qTeller does. We map reads against the whole genome, not gene models. Note that as mapping and abundance-counting algorithms continue to evolve, the algorithms we use may also change over time. Please see Data Sources for more information, including the limitations of such data in your analyses.
What parameters does MaizeGDB qTeller use to map and count reads?
We use default STAR and Cufflinks parameters, and we do not trim our fastq reads before mapping (except where noted).
Who created qTeller?
qTeller was created by Professor James Schnable of the University of Nebraska-Lincoln (Schnable Lab Website) while he was a graduate student at UC Berkeley in Mike Freeling's lab. James was generous enough to allow MaizeGDB to host a version of qTeller on our site.
How can I cite qTeller?
Please cite Woodhouse et al 2021, Bioinformatics (doi: 10.1093/bioinformatics/btab604). qTeller GitHub